Chapter 11 COVID-19 PCR Testing
The fundamental principles of PCR have remained the same since its inception, but the methods and performance improvements to DNA polymerases and reagents have been vastly changed along with the innovations in instrumentation over the years.
PCR (polymerase chain reaction) is a core technique used extensively in molecular biology research to amplify a specific DNA template in vitro rapidly. It enables researchers to generate significant quantities of sample DNA for a wide range of downstream laboratory and clinical applications, including cloning, genotyping, sequencing, mutagenesis, forensics, and the detection of pathogens to diagnose infectious diseases. Since being introduced in 1985, several iterations of the PCR process have been developed, including quantitative PCR (qPCR) for monitoring DNA amplification in real-time and reverse-transcription PCR (RT-PCR) for the detection of RNA, a tool that has become instrumental in viral diagnostics.
For accurate and sensitive PCR detection, AAT Bioquest offers a comprehensive portfolio of PCR reference dyes, deoxynucleotide triphosphates, double-stranded DNA-binding dyes, and fluorescent reporter dyes and non-fluorescent quenchers for the development of sequence-specific molecular beacons.
The polymerase chain reaction, or PCR, is one of the most well-known techniques in molecular biology. Replication of single-stranded DNA from a template using synthetic primers and a DNA polymerase was first reported as early as the 1970s [1,2]. Nevertheless, the PCR method as we know it today to amplify target DNA was not developed as a research tool until 1983, by Kary Mullis [3,4]. Since then, PCR has become an integral part of molecular biology, with applications ranging from basic research to disease diagnostics, agricultural testing, and forensic investigation. For his invention, Kary Mullis was awarded the Nobel Prize in Chemistry in 1993.
11.1 Polymerase Chain Reaction (PCR)
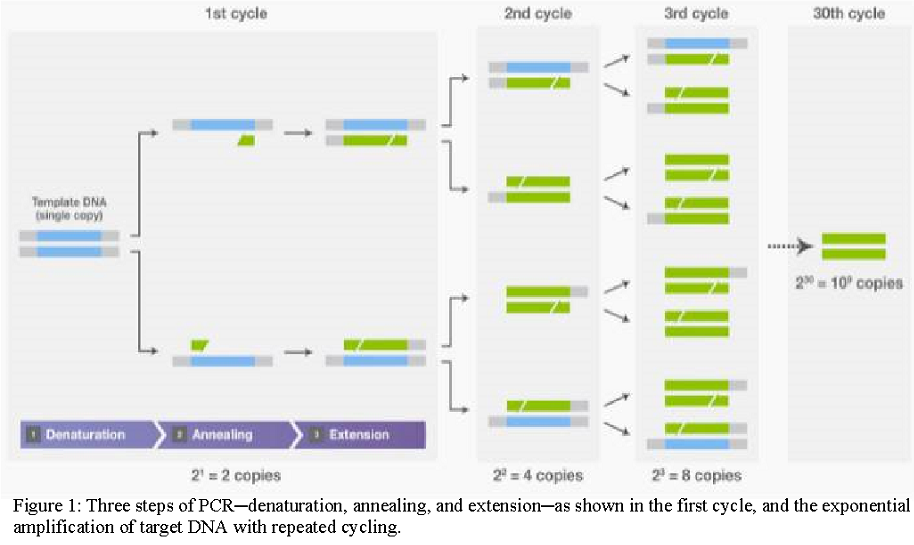
PCR is a biochemical process capable of amplifying a single DNA molecule into millions of copies in a short time. Amplification is achieved by a series of three steps: (1) denaturation, in which double-stranded DNA templates are heated to separate the strands; (2) annealing, in which short DNA molecules called primers bind to flanking regions of the target DNA; and (3) extension, in which DNA polymerase extends the 3′ end of each primer along the template strands. These steps are repeated (“cycled”) 25–35 times to exponentially produce exact copies of the target DNA (Figure 1).
11.2 DNA polymerases:
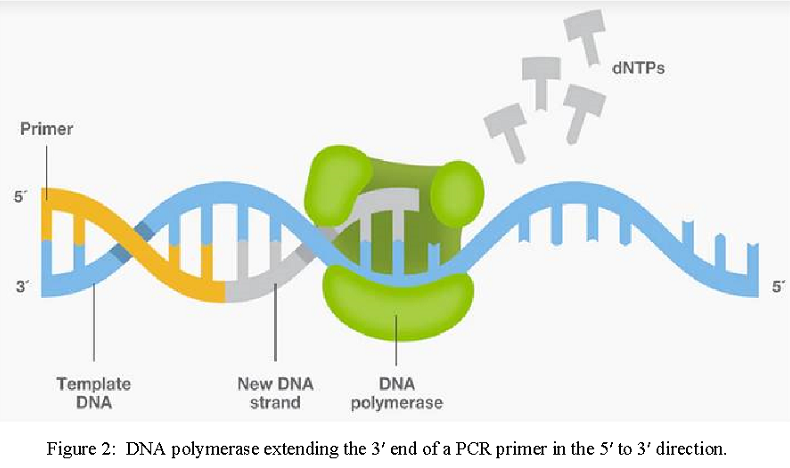
DNA polymerases are critical components in PCR, since they synthesize the new complementary strands from the single-stranded DNA templates. All DNA polymerases possess 5′→ 3′ polymerase activity, which is the incorporation of nucleotides to extend primers at their 3′ ends in the 5’ to 3’ direction (Figure 2).
In the early days of PCR, the Klenow fragment of DNA polymerase I from E. coli was used to generate the new daughter strands [3]. However, this E. coli enzyme is heat-sensitive and easily destroyed at the high denaturing temperatures that precede the annealing and extension steps. Thus, the enzyme needed to be replenished at the annealing step of each cycle throughout the process.
The discovery of thermostable DNA polymerases proved to be an important advancement, opening tremendous opportunities for the improvement of PCR methods by enabling longer-term stability of the reactions. One of the best-known thermostable DNA polymerases is Taq DNA polymerase, isolated from the thermophilic bacterial species Thermus aquaticus in 1976 [5,6]. In the first report in 1988 [7], researchers demonstrated Taq DNA polymerase’s retention of activity above 75°C, making continuous cycling without manual addition of fresh enzyme possible, and thus enabling workflow automation. Furthermore, compared to E. coli DNA polymerase, Taq DNA polymerase produced longer PCR amplicons with higher sensitivity, specificity, and yield. For all the aforementioned reasons, Taq DNA polymerase was named “Molecule of the Year” by the journal Science in 1989 [8].
Although Taq DNA polymerase significantly improved PCR protocols, the enzyme still presented some drawbacks. Taq DNA polymerase is relatively unstable above 90°C during denaturation of DNA strands. This is especially problematic for DNA templates with high GC content and/or strong secondary structures that require higher temperatures for separation. The enzyme also lacks proofreading activity; therefore, Taq DNA polymerase can misincorporate nucleotides during amplification. Where sequence accuracy is critical, PCR amplicons with errors are not desirable for cloning and sequencing. In addition, the error-prone nature of Taq DNA polymerase contributes to its inability to amplify fragments longer than 5 kb in general. To overcome such shortcomings, better-performing DNA polymerases are continually being developed to harness the power of PCR across a variety of biological applications (learn more about DNA polymerase characteristics).
11.3 Principles of PCR
PCR is commonly used to amplify a single DNA template into millions or billions of identical copies in vitro. A typical amplification reaction requires a DNA template, thermostable DNA polymerase, forward and reverse primers, deoxynucleotide triphosphates (dNTPs), and a reaction buffer (Table 1). The components are combined in a PCR or Eppendorf tube and then placed in a thermal cycler to facilitate the amplification process. While inside the thermal cycler, the PCR mixture undergoes a series of temperature and time adjustments which includes three key steps: (1) template denaturation, (2) primer annealing, and (3) primer extension.
11.3.1 PCR Process
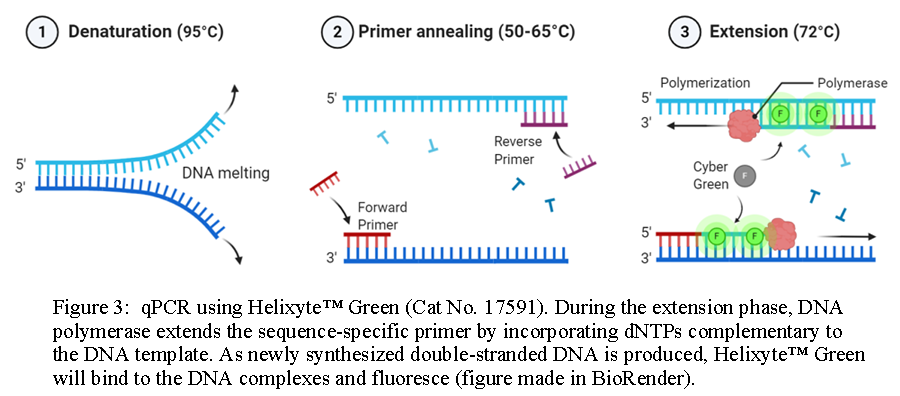
During the denaturation process, the double-stranded DNA template is heated at 95°C for two minutes. The high temperature causes hydrogen bonds between the complementary base pairs in the DNA template to separate into two single-stranded components. Next, the temperature is reduced to 50-65°C for approximately 20-40 seconds to facilitate primer annealing to each single-stranded DNA. After annealing, the temperature is increased to 70-74°C to initiate elongation. In this step, DNA polymerases will bind to and extend the primer to form a nascent DNA strand. It does so by moving along the DNA template base by base in the 5’ to 3’ direction and adds the corresponding complementary dNTP from the reaction mixture. Altogether, these three steps are referred to as a PCR cycle, and after each cycle, the number of double-stranded DNA fragments doubles. PCR cycles are repeated 25 to 30 times to amplify the original DNA template exponentially.
Table 1. Summary of the components required for PCR.

11.4 Conventional Analysis of PCR Products:
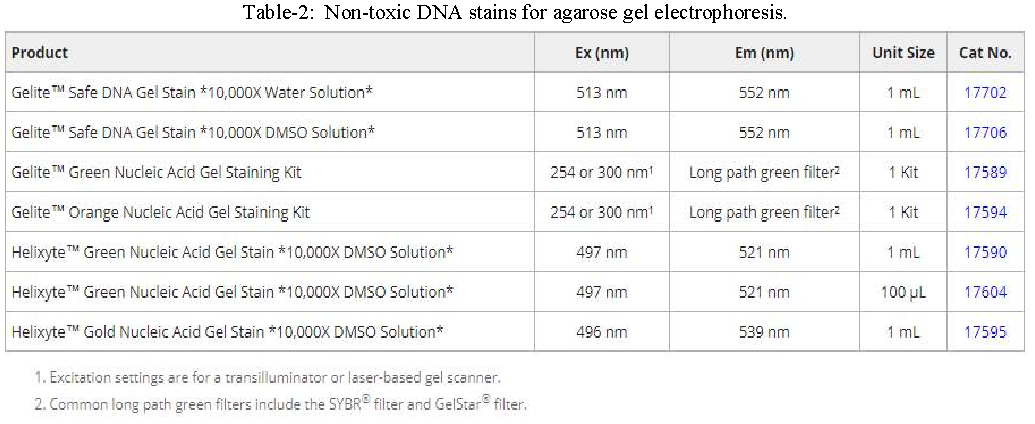
Once the PCR process is complete, the reaction products are stained with ethidium bromide (EtBr) and analyzed by agarose gel electrophoresis to determine the size and concentration of the DNA molecules. While ethidium bromide is the most commonly used dye for visualizing DNA, it is mutagenic and highly toxic through inhalation. Therefore, consider using non-toxic alternatives such as Helixyte™ Green (Cat No. 17590), Helixyte™ Gold (Cat No. 17595), Gelite™ Green (Cat No. 17589), or Gelite™ Orange (Cat No. 17594).
Types of PCR:
Conventional PCR methods, like the process aforementioned, can only amplify DNA and requires agarose gel electrophoresis to determine PCR success from the end-point of the reaction. This process is very time-consuming and is hindered by various caveats, including low sensitivity, low resolution, poor precision, non-automation, post-PCR processing, and a short dynamic range. To address these concerns, several iterations of the PCR process have been developed, including quantitative PCR (qPCR) for monitoring DNA amplification in real-time and reverse-transcription PCR (RT-PCR) for the detection of RNA, a tool that has become instrumental in viral diagnostics.
Reverse-Transcription PCR:
Reverse-transcription polymerase chain reaction (RT-PCR) is a highly sensitive technique for the detection and quantitation of mRNA expression levels. In RT-PCR, the RNA template is reverse transcribed into complementary DNA (cDNA), using reverse transcriptase. The cDNA is then used as a template for exponential amplification using standard PCR procedure (denaturation, annealing, and elongation). RT-PCR is used in various applications, including gene expression analysis, microarray validation, pathogen detection, and disease research.
Quantitative PCR:
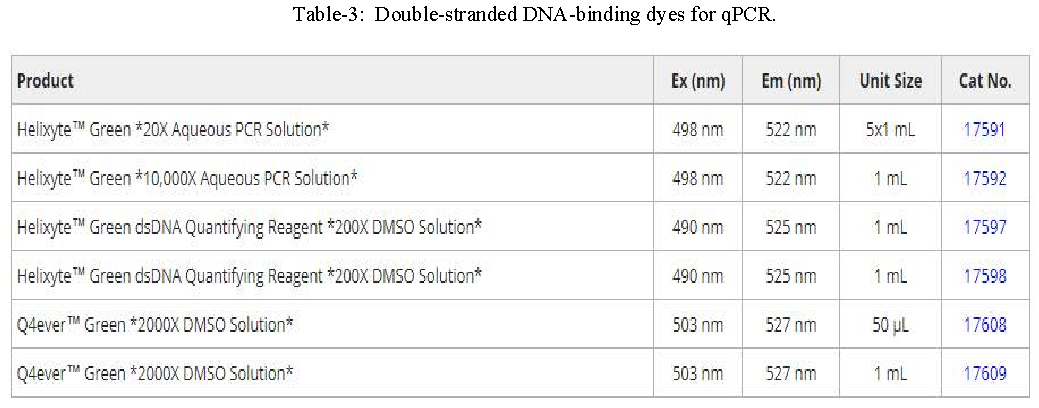
Quantitative or real-time PCR (qPCR) enables researchers to monitor the amplification of a DNA template in real-time and not at its end-point, as in conventional PCR. It does so using fluorescent reporter molecules which bind to and detect products generated during each cycle of the PCR process. As the reaction proceeds, fluorescence increases due to the accumulation of the PCR product with each amplification cycle. These fluorescent reporter molecules include dyes that bind to the double-stranded DNA (dsDNA), such as Helixyte™ Green (Cat No. 17591) and Q4ever™ Green (Cat No. 17608), or fluorescently labeled sequence-specific probes, such as TaqMan® probes, Molecular Beacons and Scorpion® probes.
Dye-Based qPCR:
In qPCR, dsDNA binding dyes are frequently used as fluorescent reporters to measure gene expression. The fluorescence of the reporter dye increases as the product accumulates with each successive cycle of amplification. Recording the amount of fluorescence emission at each cycle makes it possible to monitor the PCR reaction during the exponential phase. Compared to microarrays, qPCR is more sensitive at detecting modest changes in expression levels, making it well-suited for investigating small subsets of genes. Although dsDNA-binding dyes provide the most convenient and cheapest option for qPCR, the principal drawback to intercalation-based detection of PCR product accumulation is that both specific and nonspecific products generate signals.
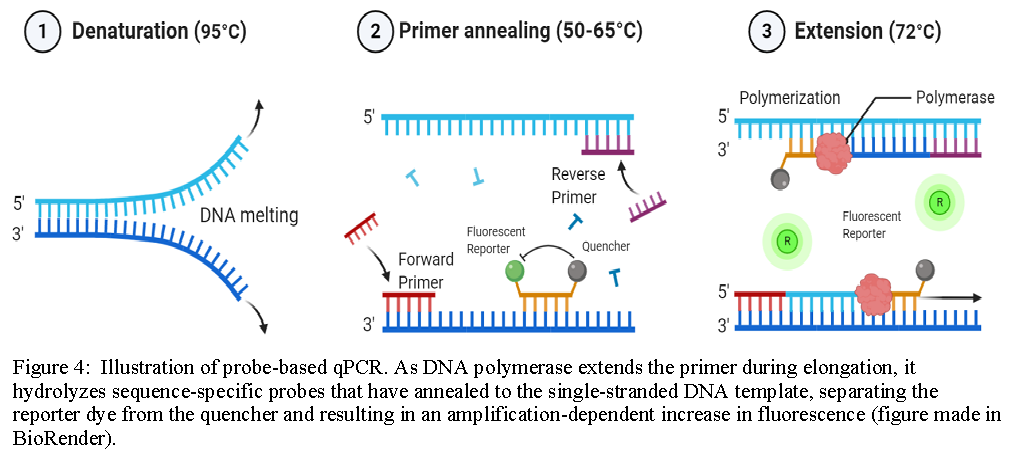
Probe-Based qPCR:
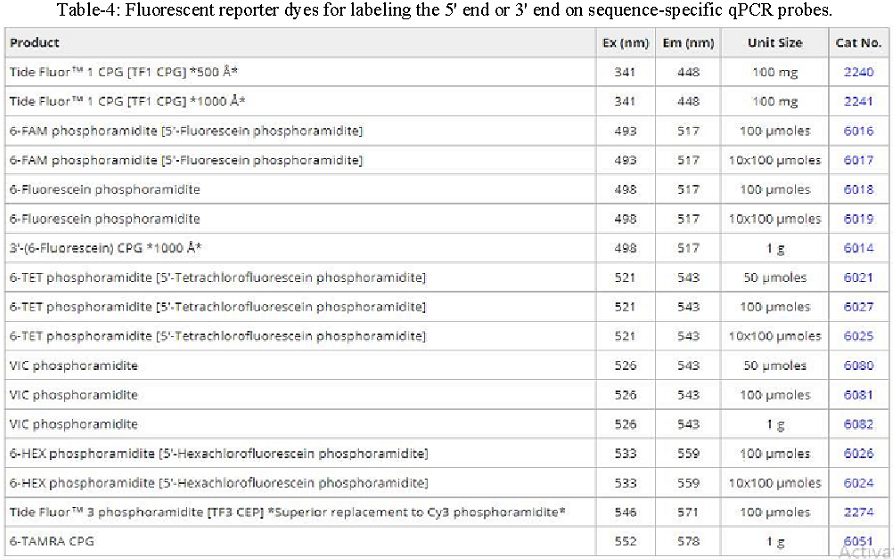
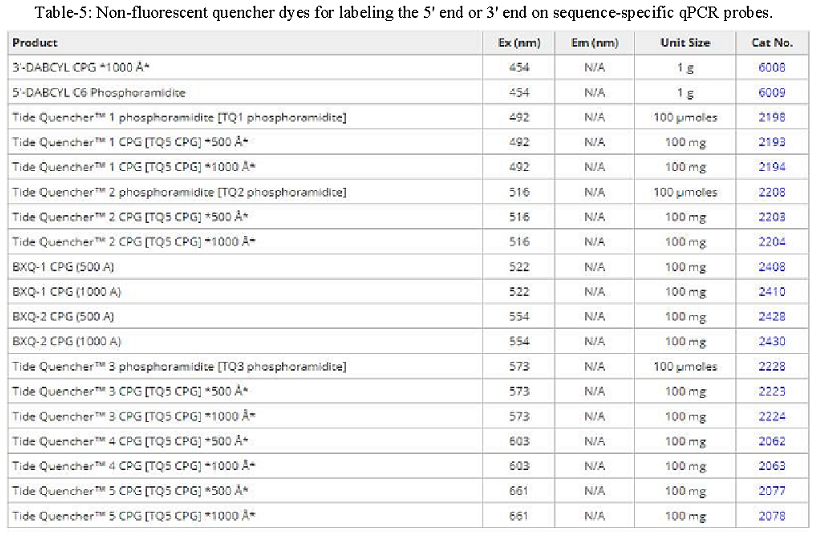
Probe-based qPCR utilizes target-specific probes to precisely measure DNA amplification at each cycle of the PCR reaction. While probe designs may vary, they typically share three key elements: a short oligonucleotide that is complementary to the target sequence, a fluorescent reporter dye-labeled to the 5’ end, and a quencher dye on the 3’ end (for information on recommended FRET pairs, see table below). Due to the biochemical phenomenon known as Förster resonance energy transfer (FRET), the fluorescence of the reporter dye is masked by the quencher dye while the probe remains intact. As DNA polymerase extends the primer during elongation, it also hydrolyzes sequence-specific probes that have annealed to the single-stranded DNA template, separating the reporter dye from the quencher and resulting in an amplification-dependent increase in fluorescence. Probe-based qPCR is a favorable method beneficial for specific hybridization, no false positives, and multiplex analysis of multiple target sequences in a single reaction tube.
References:
Kleppe K, Ohtsuka E, Kleppe R et al. (1971) Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases. J Mol Biol 56(2):341–361.
Panet A, Khorana HG (1974) Studies on polynucleotides. The linkage of deoxyribopolynucleotide templates to cellulose and its use in their replication. J Biol Chem 249(16):5213–5221.
Saiki RK, Scharf S, Faloona F et al. (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230(4732):1350–1354.
Mullis KB, Faloona FA (1987) Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155:335–350.
Brock TD, Freeze H (1969) Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J Bacteriol 98(1):289–297.
Chien A, Edgar DB, Trela JM (1976) Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J Bacteriol 127(3):1550–1557.
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239(4839):487–491.
Guyer RL, Koshland DE Jr (1989) The Molecule of the Year. Science 246(4937):1543–1546.
Smithsonian Institution Archives. The history of PCR (RU 9577)